アヘンは人類史上最古といってよい生薬であるが、痛みを和らげ眠りを誘う唯一無比の薬として、歴史的に常に脚光を浴びてきた。ルネッサンス以降に勃興した近代科学が重要な生薬の薬効成分を解明しようとしたとき、真っ先にアヘンを選んだのも歴史的必然であったといえよう。モルヒネはそのアヘンの薬効成分であり、天然有機化合物として最初に純粋な形で単離された(Serturner, 1806年)が、その平面構造が明らかにされるのに120年、絶対構造の決定まで150年を要している。分子量が300に満たない小さな化合物にもかかわらずその構造決定にこれほどの時間を要したのは、今日的視点から見てもモルヒネの構造がきわめてユニークかつ複雑だったほかに、その構造決定の基盤となる有機化学の発展を必要としていたからである。そのため、構造決定には想像を絶する時間と膨大な数の人的資源の投入が必要であった。逆にいえば有機化学の発展はモルヒネによって牽引されたということができるのである。ここでモルヒネの構造決定の歴史を記述する理由として3つ挙げておこう。一つは構造決定に参画した化学者はいずれも当代第一線の学徒であり、そのたゆまぬ努力への感謝を評するためであり、このような機会を除けば滅多にその業績は引用されることはないからである。第2は、モルヒネの構造決定が全くゼロから出発しており、古典的な構造決定法の中でもっともすばらしいロジックを提供するからである。第3は、多様な化学反応が用いられており、その過程を追跡することにより有機化学の発展の歴史を体現できるからである。
今日では、どんな複雑な天然有機化合物であってもシグナルが重複さえしなければ2D-NMRなど高度な分光学的手法を用いれば構造解析は可能である。過去に蓄積されたデータとの比較、引用によってスペクトルデータを取得した時点で対象化合物の構造のかなりの部分をイメージできているはずである。あるいは、HMBC(Heteronuclear Multiple Bond Coherence)、HMQC(Heteronuclear Multiple Quantum Correlation)を組み合わせて用いれば、未知の骨格でも導き出すことは可能である。初期、中期のモルヒネ構造決定の過程にはそれが全くないことに留意しなければならない。したがって、以降の構造決定の歴史を辿るとき、構造や反応機構に対する先入観を排して考察しなければ先人の努力を体感できないことを指摘しておく。
前述したように、モルヒネを最先端の分光学的手法を用いれば、多分、1週間ぐらいで平面構造を提出できるだろう。黎明期の有機化学が120年かかったことが今日ではたった1週間である。しかし、それが輝かしい有機化学の発展をもたらした功績を重く受けとめる必要がある。今日、我々がごく短い時間で構造解析できるのも分光学の進歩のおかげであり、決して天然物化学の進歩によるものではないからだ。今後、天然物化学を展開することで如何なる付加価値を生み出せるか、その真価が問われることになるだろう。この総説を読み終えたあと、天然物化学に従事するものであれば誰もがそう感じるはずだ。
1.モルヒネの分子量の確定とモルヒネとコデインの構造相関
今日では、大部分の有機化合物の分子量は高分解能質量分析法で決定されているだろう。19世紀の初期では、一つの有機化合物の元素分析を行うだけで立派な学術論文として認められた。無論、結果が正しいことが条件であるが、それも一定範囲内の再現性ある数値であること、また組成式が単純でありさえすればよかった。この時代で、最初にモルヒネ(1: Morphine)の分子式を提出したのはLiebigで、1831年、C34H36N2O6と発表した。1838年、RegnaultはC35H40N2O6とLiebig式よりやや大きめの分子式を発表している。一方、Laurentは、1847年、モルヒネの分子式と一致するC17H19NO3を提出している。Liebigの提出した元素分析値はC: 71.81%; H: 6.38%、RegnaultはC: 71.90%; H: 6.89%であり、正しいモルヒネの分子式から計算された値C: 71.56%; H: 6.71%と比べても、±0.4%以内に収まっている。しかし、元素分析値から正確な分子式を導くには不十分であり、正確な分子量が必要となる。1897年、von Klobukowは氷酢酸溶液の凝固点降下法によってモルヒネの分子量を285と決定し、これによってモルヒネの分子式としてLaurent式が正しいことがわかったのである。
モルヒネに次ぐアヘンの重要成分コデイン(2: Codeine)もモルヒネの精製過程の改良研究の途上で発見された。結果的には、モルヒネの構造研究でコデインは両輪の一つとして重要な役割を果たしているので、ここで述べておきたい。Serturnerが初めてモルヒネの精製に成功した後も、モルヒネの精製法を改良するため多くの化学者が参画した。Gregoryもその一人であり、1833年、アヘン微末の飽和水溶液を濃塩化カルシウム液で処理後、ろ過、濃縮して得た粗モルヒネを再結晶し、今日でいう塩酸モルヒネ(当時はGregory saltと称した)を得て、新しい精製法として発表した。当時のドイツ薬学会はその追試をRobiquetに依頼した。RobiquetはGregory法を追試したのだが、予想したよりモルヒネの収量が30%ほど少ないのに気づいた。Gregoryから提供されたアヘンを用いた場合でも大差なく、モルヒネ以外の物質が含まれていると判断したのであった。1KgのGregory saltを用いてアンモニア水で処理し、モルヒネを遊離させた(当時、モルヒネが含窒素塩基性物質であることはわかっていたので塩基で処理するのは定法であった)後、母液を濃縮したところ、固体物質を得た。この物質に水酸化カリウム溶液を処理させると、未知の塩基性物質を得ることができ、Robiquetはコデインと命名した(1833年)のである。1843年、GerhardtはRobiquetの得た物質の分子式をC18H21NO3・H2Oと決定したが、当時、モルヒネの正しい分子式が提出される前であり、コデインとモルヒネの構造上の関係は明らかではなかった。
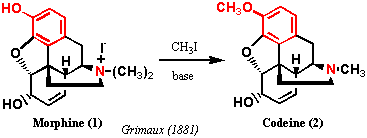
1881年、Grimauxはモルヒネの4級塩とヨウ化メチルを塩基で処理したところ、コデインと物理恒数(旋光度も含めて)が一致する物質を得た。モルヒネは強塩基中では不安定で、塩化第二鉄で呈色するが、コデインは塩基中で安定、塩化第二鉄で陰性であった。このことから、モルヒネはフェノール性水酸基をもち、コデインはそのメチルエーテルであることがわかった。実は、Robiquetはモルヒネが石灰水に溶けることを1827年に観察していたのであるが、それがフェノール性水酸基の存在によるものだということに気づいていなかった。また、粗モルヒネを水酸化カリウム溶液で処理したとき、モルヒネがその操作でなぜ除去されたか考察を怠っていた。Grimauxが前述のメチル化反応を行ったとき、Robiquetの実験からこのことに気づいていたと思われる。これに関連して無視できない知見がある。Grimauxより前の1870年代に、Wrightはモルヒネを無水酢酸あるいは酢酸クロリドで処理するとジアセテート(ジアセチルモルヒネDiacetyl morphine、別名ヘロインHeroine)を与えること、一方、コデインではモノアセテートを生成することを明らかにしており、モルヒネには二つの水酸基が存在することが示唆されていた。モルヒネの3つの酸素のうち、2つは水酸基であること、もう一つはメチル化もアセチル化も受けない不活性なエーテルであると推定された。Serturnerがモルヒネを単離してから70年以上経過したが、この時点で、明らかになったのは左上図で赤い部分だけであり、コア骨格の形は全く検討がつかない状態であった。
2.モルヒネの構造決定―フェナンスレン骨格の存在について
モルヒネ(1)の構造決定を本格的に着手したのは、1881年、von Gerichtenであったが、今日の有機化学の基準からすれば実に大胆かつ過酷な条件を用いた。すなわち、モルヒネと10倍量の亜鉛末の混合物を300℃まで加熱し、反応分解物を蒸留し精製した。その結果、得られたのはフェナンスレン(Phenanthrene)であった。しかし、用いた反応条件があまりに過酷であり、モルヒネ分子中にフェナンスレン骨格が存在すると結論づけるほどvon Gerichtenは自信をもてなかったようだ。そこで彼は、1886年、より温和な条件でモルヒネの分解反応を試みた。
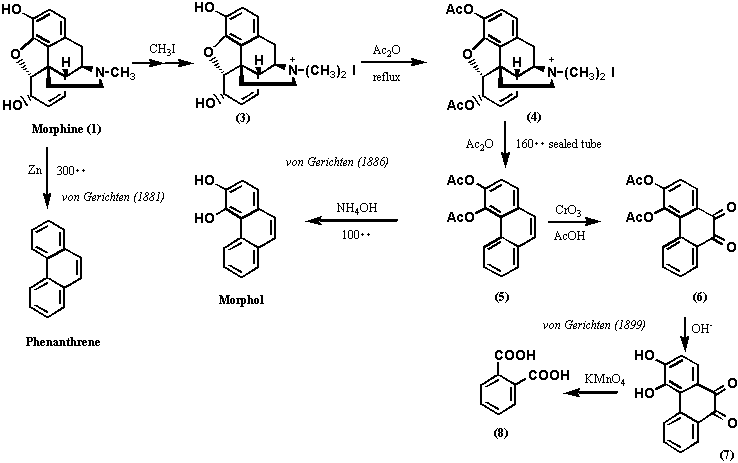
まず、モルヒネのヨー化メチル4級塩(3)を無水酢酸中で還流して得たもの(今日流にいえばジアセチルモルヒネヨー化メチル塩)(4)を封菅中で160℃に過熱して分子式C18H14O4(結果的にはジアセチルフェナンスレンDiacetylphenanthrene)で表わされる物質(5)を結晶として得た。更に、アンモニアで処理するとアセチル基が2個失われたC14H10O2で表わされる物質が得られvon Gerichtenはこれをモルフォール(Morphol)と命名した。先ほどの過酷な反応で得られたフェナンスレンと同じ炭素数なのでvon Gerichtenはこの物質がジヒドロキシフェナンスレン(Dihydroxyphenanthrene)と認識していたのは間違いないが、水酸基の位置については知らなかった。いずれにせよ、モルヒネがフェナンスレン骨格をコアに含むのは確実であり、モルヒネの炭素数は17個だから残りの3つの所在を明らかにすればよいことになった。von Gerichtenは、1883年、Barth and Weidelがモルヒネのアルカリ分解物からプロトカテキン酸(Protocatechuic acid; 3,4-Dihydroxybenzoic acid)を得ていることに気づき、アセチル基がオルト位に位置すると結論づけた。2つのアセチル基がフェナンスレンのA環に存在するは、13年後の1899年になって明らかにされた。すなわち、von Gerichtenはジアセチルフェナンスレン(5:Diacetylphenanthrene)を酸化クロムで処理してC18H12O6の生成物(6)を得たが、この物質はアセチル基は失われておらず、2個の水素原子を失ったかわりに2個の酸素原子を得ていることがわかった。この物質はオルトジアミノベンゼンと反応して鮮やかな色素(結果的にはピラジン誘導体である)を生成するので、オルトキノンであることが証明された。これによって、アセチル基はフェナンスレンの9、10位以外の位置にあることがわかった。更に、オルトキノンを加水分解し、過マンガン酸カリウムで酸化分解したところ、フタル酸(8: Phthalic acid) を得た。以上の結果は2個のアセチル基がフェナンスレンのA環に存在することを示唆するものであった(以上は図2に示す)。1882年、von Gerichtenはコデインヨウ化メチル塩(9)を酸化銀で4級アンモニウムヒドロキシド(10)に変換した後、熱分解する反応を行っている。この反応の原型は、1881年、Hofmannによって開発され今日でもホフマン分解としてアルカロイドの分解反応の定法として知られる。この反応は進行したが、化合物11では窒素は失われていなかった。そこで再び4級塩として同じ反応を試み、化合物14 (C15H10O2)を得た。一方、化合物11を無水酢酸中で160℃から180℃に加熱したところ、化合物16 (C15H12O2)を得た。元素分析の結果は化合物14と化合物16がよく似た化合物であることを示し、亜鉛末還元でいずれもフェナンスレンを精製するので、両化合物ともフェナンスレン誘導体であることは確かであった。化合物16はMorpholにメチル基が一つ多いので、Methylmorpholと命名された。一方、化合物14はMethylmorphenolと命名されたが、フリーのフェノール性水酸基はなく、以上の命名は非常に紛らわしい。以上の実験は、1889年、Knorrによって更に詳細に検討されている。化合物14と化合物16の関係については、von Gerichtenは1898年に化合物14をエタノール中で金属ナトリウム処理すると化合物16に変換されることを発見している。
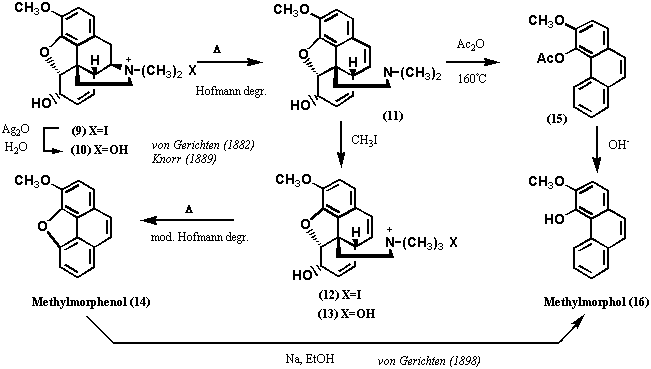
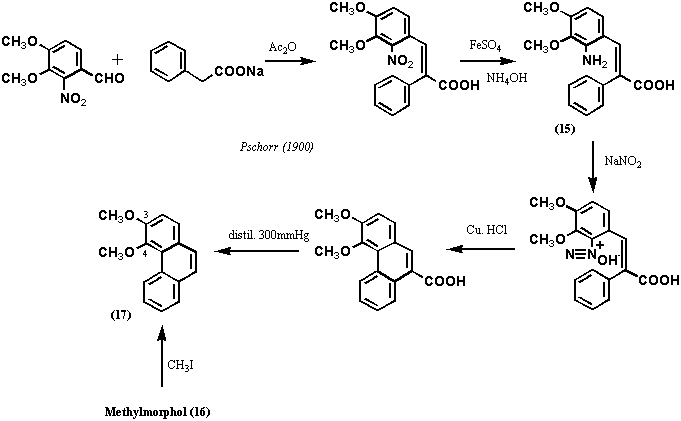
しかしながら、この時点ではまだいずれの化合物の構造も明らかではなかったが、その構造は、1900年、Pschorrが図3に示すスキームで3,4-ジメトキシフェナンスレン(17: 3,4-Dimethoxyphenanthrene)を合成(実際にはこの他に多くの誘導体を合成した)し、これとMethylmorphol (16)をメチル化したものが同定一致したことから確定した。この方法は今日でもPschorrフェナンスレン合成法としてよく知られる。von GerichtenおよびPschorrによる分解反応および化学合成によってモルヒネ(そしてコデイン)の14個の炭素がフェナンスレン骨格を形成し、A環の3位にフェノール性水酸基(メトキシ基)、4位にエーテル環を形成する酸素官能基、部分的に還元されたC環の存在が明らかにされ、モルヒネの構造決定は飛躍的に進展したといえる。
3.モルヒネ第3の酸素はどこに?
モルヒネ、コデインの3つの酸素のうち2つの場所は特定できたが、3つ目の酸素はvon Gerichtenの分解反応で失われているので、C環にアルコールとして存在すると考えられる。1903年、Ach and Knorrはコデイン(2)を過マンガン酸カリウムまたは二クロム酸カリウムで酸化するケトン体が得られることを明らかにし、これをコデイノン(17: Codeinone)と命名した(右図)。
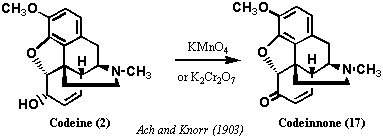
コデイノンのケトンの存在はヒドロキシルアミンと反応させるとオキシムを生成すること、またコデイノンを還元するとジヒドロコデインを与えることから酸化反応の間に骨格の転位は起きていないことが明らかとなった。この段階ではアルコールの位置についてはまだ特定できなかった。Freundは、1897年、von Gerichtenの分解反応をコデイノンについて行った。すなわち、コデイノンを無水酢酸中還流して化合物18(モノメトキシジアセテート)を得た。元素分析の結果ではコデイノンの3つの酸素官能基を全て含んでいた。この化合物18をアルカリ加水分解し、ヨー化メチルでメチル化し、トリメトキシフェナンスレン(20) とした後、酸化分解したところ、モノメトキシフタル酸を生成した。モノメトキシフタル酸はPschorrによりm-メトキシフタル酸 (21: m-Methoxyphthalic acid) と同定された。これによってトリメトキシフェナンスレン(20)の第3のメトキシ基は6位または7位であることがわかった。最終的には、1902年、Pschorrが合成した3,4,6-トリメトキシフェナンスレンがコデイノンの分解で得られたトリメトキシフェナンスレンと完全に一致したことで、6位と決定された。
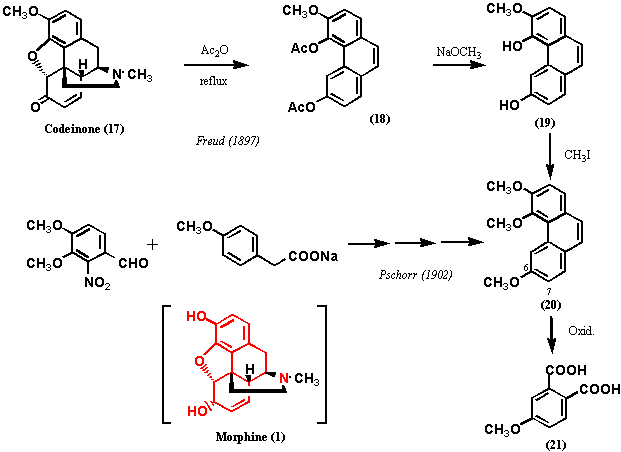
以上、モルヒネの14個の炭素はフェナンスレン骨格を形成し、3位にフェノール性水酸基、4位と5位はエーテルブリッジで結合され、6位にアルコール性水酸基が存在することが明らかにされたのである。モルヒネが単離されてから約100年後、Wright、Grimaux、von Gerichten、Pschorrら当代一流の学徒の業績によりやっとここまで到達したのである。
4.モルヒネ分子内のエチルアミンブリッジの存在
前述したように、1889年、Knorrはコデインの4級アンモニウムヒドロキシド(10)についてホフマン分解反応を行い、分解産物(12)を得ているが、窒素原子は残存していた。そこで、再び4級アンモニウムヒドロキシド(13)とし分解反応したところ、前述したように化合物14(結果的にはフェナンスレン誘導体)を得た。この時、Knorrは揮発性成分の捕捉を試み、トリエチルアミン(23)を金塩(C3H10NAuCl4)および白金塩(C6H2ON2PtCl6)として得ていた。この結果から、Knorrはトリエチルアミンの二つのメチル基はメチル化で導入されたものであるが、もう一つのメチル基はアルカロイドにもともと存在するものであり、コデイン(モルヒネ)にN-メチル基の存在を指摘した。この結論が正しかったことは、1914年、von Braunがジアセチルモルヒネ(25: Diacetyl morphine; Heroine) について、今日、von Braun反応として知られる一連の反応を行って、デメチルジアセチルモルヒネ(27: Demethyl-diacetyl morphine) を誘導したことで証明された。
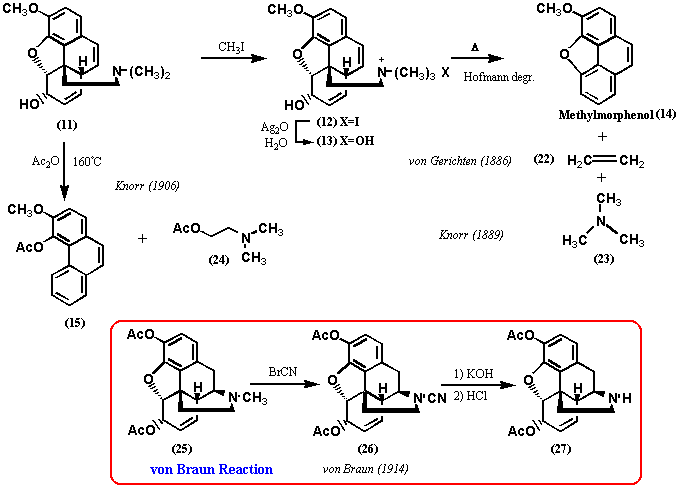
1886年、von Gerichtenは以前に自ら行った第二段階のホフマン分解反応で、非塩基揮発物を臭素付加体1,2-ジブロモエチレンとして捕捉した。Knorrは1906年、化合物11を無水酢酸中還流する分解反応を精査し、1-アセトキシ-2-(ジメチルアミノ)エタン(24: 1-Acetoxy-2-(dimethylamino)ethane) を補足したが、おそらくvon Gerichtenの結果を知った上での結論であろう。これらの結果により、モルヒネの残りの3個の炭素、1個の窒素が分解反応で捕捉されたことになるが、Knorr自身はこのフラグメントが-O-CH2CH2N(CH3)-であると結論し、末端酸素が分解反応の過程で導入されたこと、モルヒネの3個の酸素の所在が既に明らかにされていることに気づかなかった。一方、von Gerichtenも残りの部分構造を提出するには至らなかった。コデインの第一段階のホフマン分解反応の産物の構造は当時不明であったが、第二段階の反応を行うとき、4級塩を形成しているので、窒素はフェナンスレン骨格のどこかに結合していたことは確実である。つまり、窒素原子はモルヒネ分子内で複素環を形成していると考えられる。
5.エチルアミン側鎖はどこに?
以上、モルヒネを構成する全ての原子が何らかの分解産物中に捕捉された。そして、-CH2CH2N(CH3)-以外は前節で述べたようにフェナンスレン骨格と3つの酸素官能基の位置が確定しているので、この残基の両端がフェナンスレン骨格のどこに結合しているかが最後のパズルとなった。まず、窒素の結合位置について考察してみたい。
1903年、Ach and Knorrはコデインを低温下、希硫酸中クロム酸で処理すると、コデイノンとは明らかに異なる化合物28を得た。この物質は無水酢酸によってジアセチル体29を与えるので、酸化反応で新たに水酸基が生成したことを示唆する。化合物28はオキシコデイン(Oxycodeine)と命名された。1906年、Knorr and Schneiderはオキシコデインをホフマン分解に供したところ、化合物32を得たが、窒素及び第4の酸素原子を保持していた。化合物32を無水酢酸中で還流すると化合物33 (C19H16O5)を生成、この物質は2個のアセチル基をもっていた。酸化クロムで酸化すると既知化合物の化合物34を生成、2個あったアセチル基の一つは失われたので、第4の酸素官能基は9位あるいは10位に存在することが明らかとなった。したがって、オキシコデイン(28)は6位のほか、もう一つのアルコールを9位あるいは10位にもつ。ホフマン分解では二重結合の生成を伴うことはわかっているので、9、10位の炭素は飽和していることも明らかである。通常、加熱を必要とする31から32への反応が自然に起きることから、窒素が結合する飽和炭素上に水酸基が結合していることは確実である。
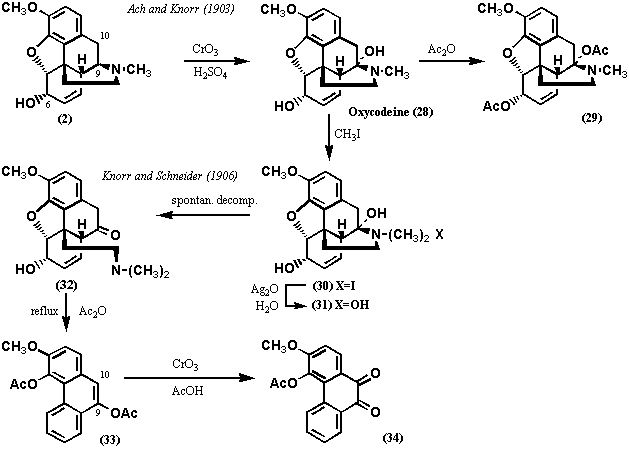
N末が9位、10位のどちらに結合しているかを区別するのは難しいが、モルヒネ(1)、コデイン(2)の分解反応でフェナンスレンの生成過程でN末が除去されるとき、隣の炭素上の水素が脱離するが、ベンジル位(10位)の水素の方がはるかに脱離しやすいので、N末の結合位置として9位の可能性の方がずっと大きいであろう。N末の結合位置は、あるモルヒネ(1)の反応生成物の構造が明らかになったことで確定した。これについては図10を参照。
1870年、Matthiessen and Wrightはコデイン(2)を塩酸中で加熱すると、4種の塩化物35a-dを与えることを発見し、これらをクロロコライド(chlorocolides; 35a-d)と命名した。
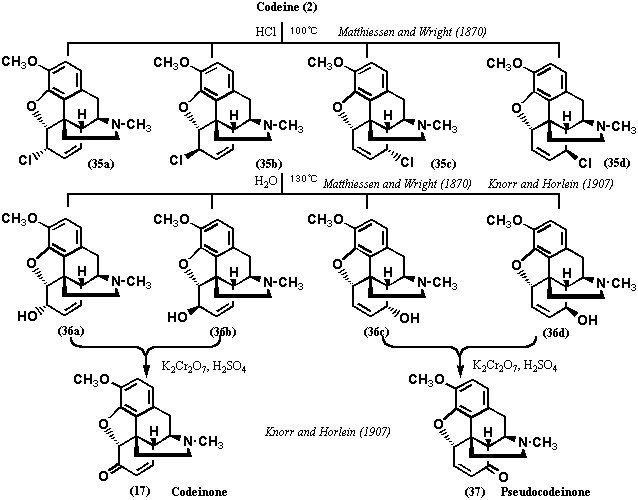
クロロコライド(35a-d)が塩化物であることは元素分析の結果から明らかであり、コデイン(2)の水酸基が塩素で置換されたものであることはクロロコライド(35a-d)がアセチル化されないことでわかった。また、クロロコライド(35a-d)は、コデイン(2)と5塩化リン(von Gerichten, 1881)、3塩化リン(Lees, 1907)、塩化チオニル(Wieland etc, 1911)で処理したとき、生成することも後に明らかにされた。無論、当時はクロロコライド(35a-d)の構造は不明であったが、分離、精製は可能であった。塩化物35a-dの混合物あるいはそれぞれの精製物を加水分解するとアルコール体を与え、その一つはコデイン(2)そのものであった。アルコール体をニクロム酸カリウムで酸化すると、2種(うち一種はコデイン)はコデイノン(17)、残りの2種(プソイドコデイン(Pseudocodeine)、アロプソイドコデイン(Allo-pseudocodeine)と命名された)はプソイドコデイノン(Pseudocodeinone; 38)と命名したコデイノンの類縁物質を与えた。これらの実験は、1907年、Knorr and Horleinが追試、確認しており、更にプソイドコデイノン(38)を無水酢酸中で加熱分解し、フェナンスレン誘導体39を得た。
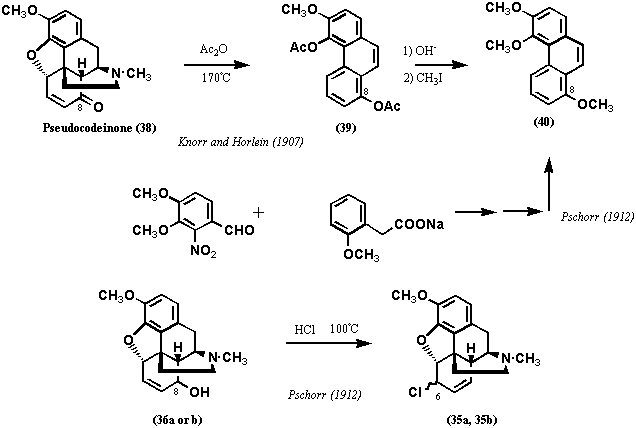
化合物39はアルカリ加水分解後、メチル化して得た化合物40は、1912年、Pschorrが合成した3,4,8-Trimethoxyphenanthrene (40)と一致した。これらの結果から、プソイドコデイノン(38)は8位に酸素官能基をもつコデイノン(17)の位置異性体であることが明らかとなり、またプソイドコデイン、アロプソイドコデインがコデインの位置異性体であることもわかった。更に、プソイドコデイン、アロプソイドコデイン(36a, b)を塩酸中で加熱すると、塩化物の混合物35a, bに戻るので、この反応の途上で転位反応は起きていないことは明らかである。結局、Matthiessen and Wrightの実験結果は、コデイン(2)のアルコール水酸基と塩素イオンとの間のSN1/SN1’反応による置換反応であることを示すが、当時はこのような概念は全くなかった。これらの実験結果は8位に置換基がないことを示唆し、エチルアミンのC末およびN末が8位に結合していないことを示唆する。
コデイン(2)をホフマン分解して得られる化合物11を塩基処理すると異性体41が得られる(Knorr, 1894; Pschorr, 1906)。41をニッケル触媒で還元すると水素ガスは1当量だけ吸収され、ジヒドロ体42を得る。一方、化合物11を同じ条件で還元すると、1当量の水素ガスを吸収、ジヒドロ体43を得るが、化合物42とは一致しなかった(von Gerichten, 1899; von Braun, 1927)。しかし、化合物43はジヒドロコデイン(44: Dihydrocodeine) をホフマン分解して得られるものと一致した(Wieland and Koralek, 1923)。
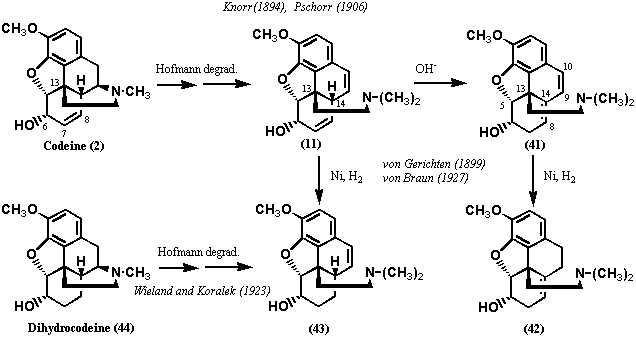
以上の実験結果は、化合物42はC環の8、14位に二重結合があり、化合物11のアルカリ処理によって14位の水素が引き抜かれ、8、14位、9、10位が共役化したことを示唆する。したがって、コデイン(2)の7、8位に二重結合が存在すること並びに14位には置換基(すなわちエチルアミンのN、C末)が存在しないことがわかる。しかし、ここで一つの疑問が起きる。すなわち、化合物41では二重結合の異性化が8、14位でストップしていることである。もし、13位に水素が結合していたなら、14位水素が引き抜かれれば、13、14位に異性化した方がB環が芳香化するので、より安定になるはずである。したがって、この実験結果は13位には水素がないこと、すなわち、C末の結合位置が13位であることを強く示唆するのである。
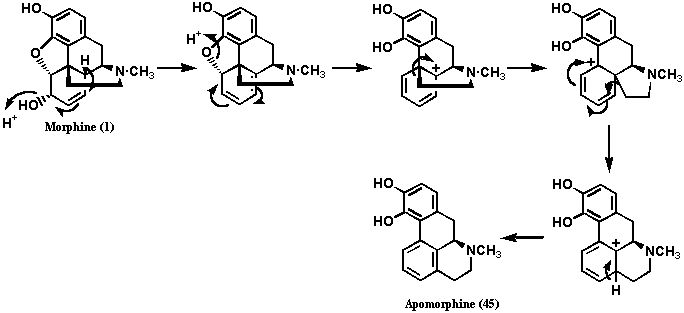
1870年、Matthiessenはモルヒネを封管中で塩酸とともに高温で処理してアポモルヒネ (45: Apomorphine)と名付けた物質を得た。元素分析によってアポモルヒネ(45)は分子式がC17H17NO2と計算されたが、モルヒネの脱水形に相当するものであった。Pschorrはアポモルヒネ(45)のフェノール性水酸基をメチル化、ヨウ化メチルで4級塩(47)としたのち、ホフマン分解を行い、塩基性化合物49を得た。化合物49についてもう一段階ホフマン分解を行ったところ、非塩基性オレフィン50を得、過マンガン酸カリウムで酸化するとカルボン酸51を得た。化合物51をクルチウス(Curtius)転位でカルボキシル基をアミノ基(化合物52)としたのち、ジアゾ化を経てフェノール53とした。化合物53をメチル化したものは既知物質3,4,8-トリメトキシフェナンスレン (40)であった。
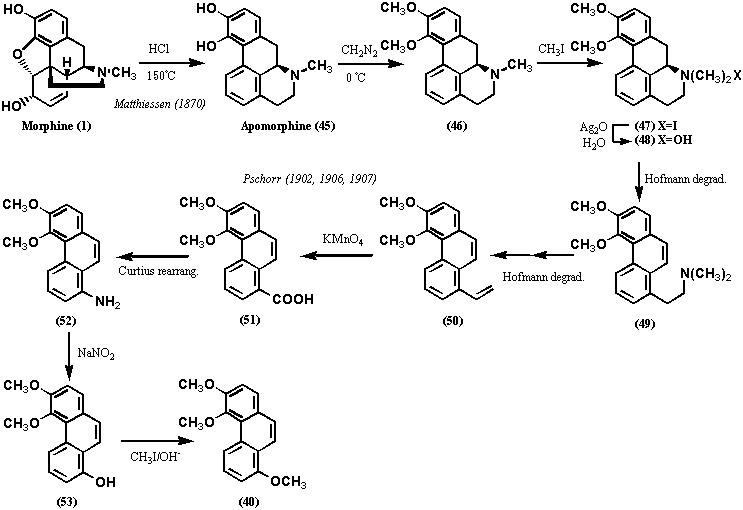
Pschorrの行った反応を逆に辿ればアポモルヒネの構造式として(45)に到達する。この構造式ではエチルアミンのC末は8位に結合し、前述の結果と矛盾するので、この反応で転位反応が起きていることは確実であるが、当時はどんな反応が起きているのか知る由もなかった。しかし、N末については、アポモルヒネ(45)が光学活性であることおよびフェナンスレン骨格のB環に結合しており、これまでの実験データと矛盾しないので転位はしていないと考えられた。したがって、N末の結合位置は、前述の推定通り、9位であることがわかった。因みに、アポモルヒネ(45)の生成メカニズムは今日では図11のスキームで進行すると考えられている。
6.Wieland and Kotake説とGulland and Robinson説
これまでの結果から、モルヒネ(1)の推定構造式としてエチルアミンのN末は9位、C末は13位に結合した構造、すなわち54bがもっとも
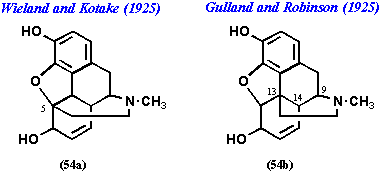
有力であることが理解できる。この構造は、1925年、Robinsonによって提出されたものであり、以上述べた論拠によるものであった。しかし、当時、著名な有機化学者によるもう一つの説があった。それはWieland and Kotakeが提唱した54aであった (右図)。今まで、述べた実験データからはこの構造式は明らかに不利なのに、なぜこの構造式が提唱されたのであろうか。Wielandとともに54aの構造を共同提案したKotakeとは後の大阪大学理学部化学科教授で、戦後のわが国の天然物化学をリードしてきた小竹無二雄(1894-1976)であった。小竹は、1975年7月、昭和薬科大学白樺湖校舎において開催された天然物化学談話会に、恩師真島利光東北帝国大学理学部教授の思いでについて語るため招かれた。天然物談話会とは、天然物化学を研究する老若の学徒が一同に会し、寝食をともにして、天然物化学に関する様々な話題について自由に語り合う会合であり、米国のGordon Conferenceをモデルにしたものであった。筆者は、この席上で小竹にモルヒネの構造論争のいきさつについて質問したことがあった。小竹自身は54aの構造式に、当初、納得していなかったが、最終的には熟慮の上でWielandが決断したという。では、Wielandが何故この構造式にこだわったのか、以下、これについて述べることとする。
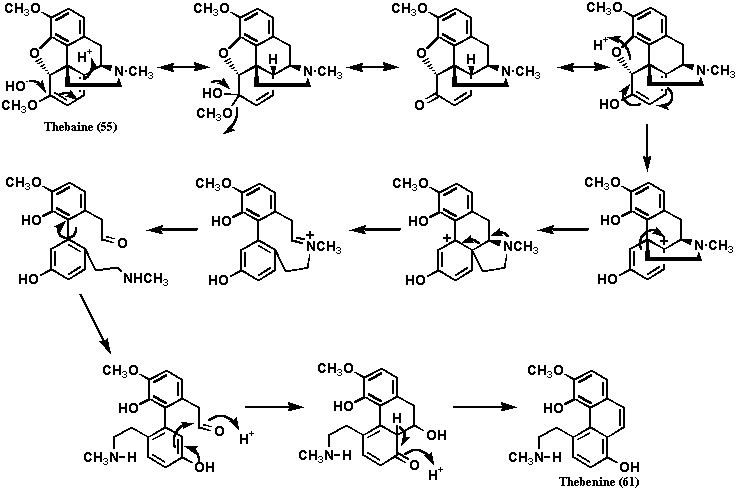
1870年、Hesseはアヘンの副成分の一つであるテバイン(55)を希塩酸中で数分煮沸して新規塩基成分テベニン(61: Thebenine) を得た。元素分析によってその分子式はC18H19NO3と計算され、テバイン(55)の分子式C19H21NO3と比べると、メチル基が1個分少ないものであった。また、同じ反応をメタノール中で行うと、テバイン(55)と同じ分子式の塩基性物質56が得られ、メテベニン(56: Methebenine) と名付けられた(図12)。1904年、Freundはジメチルテベニンのヨウ化メチル体57をホフマン分解して、トリメチルアミンN(CH3)3とともに非塩基性オレフィン58を得た。化合物56から化合物57を調製するとき、ヨウ化エチルを用い、ホフマン分解したとき、メチルジメエルアミンCH3N(CH2CH3)2が得られるので、メテベニン(56)そしてテベニン(61)が非環状の2級メチルアミンをもつことは明らかであった。したがって、メテベニン(56)は環状の3級アミンであるアポモルヒネ(45)とは構造が異なることがわかる。オレフィン58を過マンガン酸カリウムで酸化し、生成するカルボン酸59を高温で過熱すると脱炭酸が起き、既知物質である3,4,8-トリメトキシフェナンスレン(40)が得られた(Pschorr, 1910)。この実験データから、テベニン(61)を亜鉛末とともに高温処理したとき、フェナンスレンの生成が予想されたが、実際に得られたのはピレン(62: Pyrene) であった(von Gerichten, 1901; Freund, 1910)。このデータから5位にエチルメチルアミン基が結合していることが示唆された。テベニン(61)をホフマン分解したとき、モノフェノール体であるテベノール(63)が得られた。エテベニン(60: Ethebenine) (テバイン(55)をエタノール中塩酸で煮沸して得られる塩基物質)をホフマン分解したときはエテベノール(64: Ethebenol) が得られ、フリーのフェノール基は存在しなかった(塩化第二鉄試薬で発色しなかった)。このことからテベノール(63: Thebenol) はホフマン分解でエチルアミン側鎖が分解して生成したエテニル基が環化したエーテル体と推定された。以上の実験結果からテベニン(61)、メテベニン(56)のエチルアミン側鎖は5位に結合していることが決定的となり、Wieland and Kotakeはモルヒネ(1)においてもエチルアミンのC末は5位に結合していると結論したのである。当時の化学の水準ではテバイン(55)の転位反応を予見することは困難であり、5位にC末が結合していることを積極的に否定するデータはなく、Wielandの決断は決して荒唐無稽ではないことが理解できよう。Robinsonも図12に示すテバイン(55)の反応結果には相当悩んだことだろう。
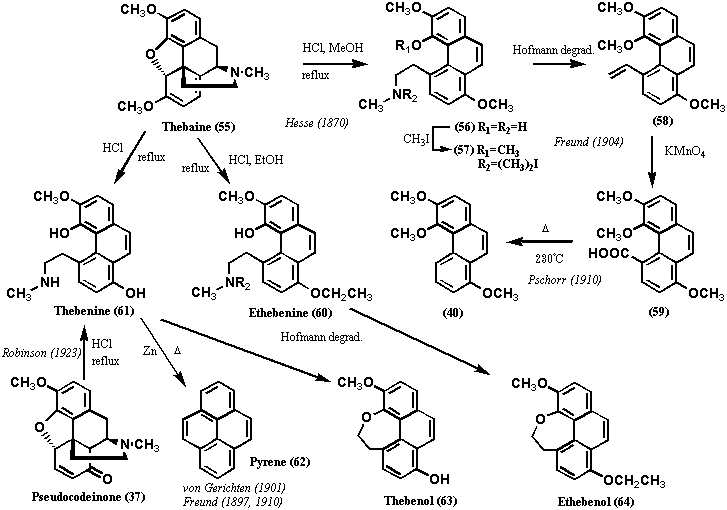
一方、アポモルヒネ(45)も、亜鉛末とともに高温処理したとき、フェナンスレンを生成しなかったのであるが、その場合は、エチルアミン側鎖のC末が8位に転位していることが明らかにされていた(もともと8位には置換基がないという実験データがあった!)。Gulland and Robinsonは、モルヒネ(1)からアポモルヒネ(45)が生成することから、モルヒネ(1)を酸処理すると未知の複雑な転位反応が起きることを知り、モルヒネの構造を推定するのに以上述べた一連の実験結果を考慮しなかったのである。テバイン(55)からテベニン(61)への転位反応のメカニズムは今日でも詳細は明らかではない。ここではその一説を挙げておくが、必ずしも有機化学的に納得できるものではないことに留意する必要がある(図13)。
7.モルヒネの構造の最終決着と天然物化学の潮流の変化
以上、モルヒネ(1)の構造に対して、Wieland and Kotake説とGulland and Robinson説の2大有力説が1925年までに提唱された。Serturnerが、1805年、モルヒネを純品として単離して以来、120年を経ていた。いずれの説も推定の域を出ず、決定的な証拠を欠いていたが、Gulland and Robinson説の方が多くの支持者を得ていたようである。この論争の最終決着は、1952年、米国ロチェスター大学教授Gatesがモルヒネの全合成を報告し、Gulland and Robinsonの構造式が正しいことが証明した。モルヒネの3次元構造式は右の図のように表される。小さな分子量ながら、その構造がきわめて複雑であることは、その分子をコンピュータ上で回転させることによって実感できる(右構造式はwebアプリのMolviewが埋め込まれているので、PCのマウスのカーソル操作(スマホ・タブレットではスワイプ)で自由自在に動かすことができる。3D構造式のサイズはマウスホイール(スマホ・タブレットではピンチイン・ピンチアウト)で調節できる。)。
Gatesの業績は天然物化学の金字塔であるが、1940年代から1960年代にかけて多くの複雑な天然有機化合物が次々と合成された時代でもあった。すなわち、有機化学がおよそどんな有機化合物でも合成できるほどまで進歩した結果であったのである。1970年代から、有機化合物の構造決定法として核磁気共鳴(NMR)がハード(超伝導マグネットを用いた高分解能装置)およびソフト(二次元NMRスペクトル)の両面で著しい進歩を遂げた。これ以降は、有機化合物の構造を決定するのに分解反応を行う必要はなくなり、また、ごく微量の試料でも構造を決定できるようになった。すなわち、有機化合物の構造決定に有機化学の知識はさほど必要としなくなったのである。こうした天然物化学の構造的な変化は、有機化学の潮流の変化として現われ、新しい有機化学反応は天然有機化合物の構造決定からではなく、その全合成を行う過程で発見されるようになった。もはや、天然有機化合物の構造決定は「化学」というより「各種分光スペクトルの解析術」となり、天然物化学者の新規化合物の探索の意欲を減退させかねない深刻な構造問題をもたらすことになった。しかし、生物の生産する二次代謝物は新薬創製のもっとも有望な「シード源」であり、地球上最大の潜在的化学物質ライブラリーである。新薬シード探索への意欲こそが、既に学問として成熟した天然物化学の唯一の生きる道ではあるまいか。
![]() 参考文献
参考文献
- G. Butora and Tomas Hudlicky, Organic Synthesis: Theory and Applications, 4, 1-51 (1998).